Drugs of Potential Interest in Haemostasis
Introduction
The following lists some of the commonly encountered drugs in haemostasis, their mode of action and how they are monitored [if necessary] in the laboratory.
| Drug | Mode of Action | Monitoring | Comments |
|---|---|---|---|
| Vitamin K Antagonists [VKAs] | |||
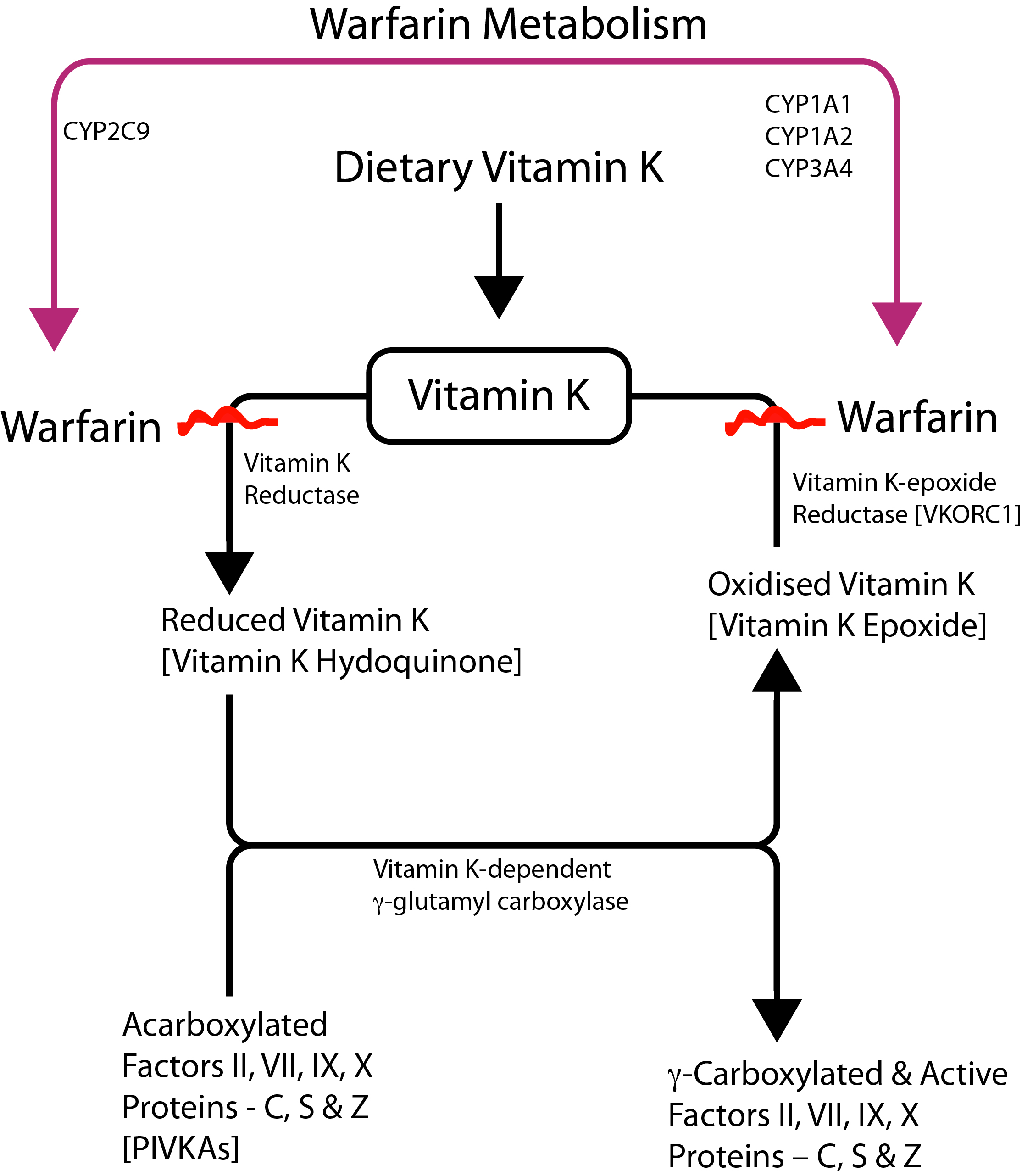
| Warfarin | Vitamin K Antagonist. Inhibits γ-carboxylation of Factors II, VII, IX and X [+ Proteins C, S and Z.] Click HERE for a diagram outlining Vitamin K metabolism and the action of warfarin |
INR | Warfarin has a T½ of 35-45 hours The inter-individual variability observed with VKAs response is in part due to genetic variability arising from mutations in the CYP2C9 [cytochrome P450 genes] and VKORC1. Mutations in CYP2C9 have been linked to decreasing activity in metabolising the VKAs leading to a prolonged T½ and over-anticoagulation. Similarly mutations in the VKORC1 gene have been linked to a decrease in requirements for warfarin. A PK algorithm has been proposed to prevent over- or under-anticoagulation - see references. VKORCI encodes the VKORC1 enzyme a small transmembrane unit of the endoplasmic reticulum and is primarily transcribed in the liver. Various polymorphisms and mutations within the VKORCI gene have been reported. The polymorphisms are associated with a reduction in the levels of VKORC1 and therefore a reduction in the amounts of warfarin that an individual requires to achieve a stable INR. Mutations within the VKORC1 gene have been associated with a reduction in the levels of all the Vitamin K dependent clotting factors. |
| Phenprocoumon | Vitamin K Antagonist. Inhibits γ-carboxylation of Factors II, VII, IX and X [+ Proteins C, S and Z.] |
INR | Phenprocoumon has a T½ of 80-270 hours. The inter-individual variability observed with VKAs response is in part due to genetic variability arising from mutations in the CYP2C9 [cytochrome P450 genes] and VKORC1. Mutations in CYP2C9 have been linked to decreasing activity in metabolising the VKAs leading to a prolonged T½ and over-anticoagulation. |
| Acenocoumarol | Vitamin K Antagonist Inhibits γ-carboxylation of Factors II, VII, IX and X [+ Proteins C, S and Z.] |
INR | Acenocoumarol has a T½ of 8-24 hours. The inter-individual variability observed with VKAs response is in part due to genetic variability arising from mutations in the CYP2C9 [cytochrome P450 genes] and VKORC1. Mutations in CYP2C9 have been linked to decreasing activity in metabolising the VKAs leading to a prolonged T½ and over-anticoagulation. |
| Tecarfarin [ATI-5923] |
Vitamin K Antagonist. Inhibits γ-carboxylation of Factors II, VII, IX and X [+ Proteins C, S and Z.] |
INR | Tecarfarin is an oral vitamin K antagonist that has been engineered so that it is not metabolised through the cytochrome P450 [CYP] pathway. For these reasons it has a decreased potential to interact with drugs that inhibit CYP450 enzymes. |
| Phenindione | Vitamin K Antagonist. Inhibits γ-carboxylation of Factors II, VII, IX and X [+ Proteins C, S and Z.] |
INR | Phenindione is an inandione derivative - rarely used due to a high incidence of adverse events including skin rashes and abnormal liver function tests. Phenindione has a T½ of 5-10 hours. |
| Anisindione | Vitamin K Antagonist. Inhibits γ-carboxylation of Factors II, VII, IX and X [+ Proteins C, S and Z.] |
INR | A synthetic inandione derivative. |
| Heparin and Heparins | |||
| Unfractionated Heparin [UFH] | Inhibits Factors IIa and Xa | 1. APTT 2. Anti-Xa assay 3. Activated Clotting Time [ACT] |
UFH is a sulphated polysaccharide with a molecular weight range of 3,000-30,000Da. It binds to Antithrombin in plasma causing a conformational change in its structure and an acceleration in its inhibitory activity. Heparin binds to AT through a high affinity pentasaccharide binding site which is present in ~ 1/3 of heparin molecules. Maximal anti-IIa activity is dependent upon the binding of heparin to both thrombin and heparin. Molecules <18 saccharide units lack the necessary chain length to form a bridge between the two molecules and so short chain heparin molecules have primarily anti-Xa inhibitory activity. UFH binds to a number of plasma proteins and which accounts for the variable intra-individual anticoagulant response. |
| LMWH | Primarily Xa inhibition but some IIa. Xa:IIa ratio varies from LMWH to LMWH |
Anti-Xa assay | LMWHs [of which there are many] are prepared from UFH and are enriched for short-chain heparin molecules and so have primarily anti-Xa activity. LMWHs have a molecular weight range of 1,000-10,000 Da with a mean range of 4500-5000 Da. LMWHs have more predictable pharmacokinetics than UFH due to reduced binding to plasma proteins and so can be given once daily without [in the majority of patients] any need for laboratory monitoring. LMWHs are excreted through the kidneys and may accumulate in patients with impaired renal function. |
| Fondaparinux | Specific Xa inhibitor | Anti-Xa assay | Fondaparinux is a synthetic pentasaccharide Xa inhibitor identical to that found in LMWH and UFH. It is given subcutaneously and has a T½ of 17-21 hours. Fondaparinux is renally excreted and so may accumulate in patients with impaired renal function. |
| Idraparinux | Idraparinux is a hypermethylated derivative of fondaparinux and binds Antithrombin with a strong affinity that accounts for its long T½. Idraparinux is a specific Xa inhibitor |
Anti-Xa assay | Idraparinux is similar to Fondaparinux but with a T½ of 80-130 hours which means the drug only requires to be administered weekly. However, although the drug is effective it may be associated with an increased risk of bleeding. Idrabiotaparinux is similar to Idraparinux but contains a biotin group which allows its anticoagulant activity to be rapidly reversed with avidin. |
| Danaparoid | Specific Xa inhibitor | Anti-Xa assay | Danaparoid is a mixture of Heparan Sulphate, Chondroitin Sulphate and Dermatan Sulphate. Danaparoid has an anti-Xa elimination T½ of ~25 hours but the thrombin generation-inhibiting activity is eliminated with a T½ of ~7 hours. Danaparoid is excreted through the kidneys and so may accumulate in patients with impaired renal function. Danaparoid has an anti-Xa:anti-IIa activity ratio of 20 compared to ~2.5 for the LMWHs and 1 for UFH. |
| Anti-platelet Agents | |||
| Aspirin | Aspirin irreversibly inhibits platelet cyclooxygenase [COX1] - a key enzyme involved in the generation of Thromboxane A2 [TxA2]. | Aspirin has a relatively short half-life of 3 hours but its effects on platelets is irreversible and so the effects last for the life-span of the platelet [7-10 days] The VerifyNow system is a whole blood method that has been evaluated as a rapid method for establishing the anti-platelet efficacy of aspirin therapy. |
Aspirin inhibits platelet COX1 and as platelets are non-nucleated they are not able to re-synthesise this. However, although COX1 is found in endothelial cells, these cells are nucleated and so can re-synthesise the enzyme. Aspirin therefore inhibits platelet aggregation through the inhibition of TxA2 generation but has no effect on endothelial cell prostacyclin [PgI2] production. |
| Clopidogrel | Binds to the P2Y12 receptor preventing ADP-induced platelet aggregation. ADP binds to two receptors - P2Y1 and P2Y12 but the P2Y12 receptor is the primary receptor involved in ADP-stimulated platelet activation of the GpIIb/IIIa receptor. |
Clopidogrel has a T½ of ~ 7-8 hours. The VerifyNow system is a whole blood method that has been evealuated as a rapid method for establishing the anti-platelet efficacy of aspirin therapy. |
Clopidogrel belongs to a group of drugs known as the thienopyridines of which ticlopidine was the first member to be used therapeutically. Ticlopidine was associated with a number of haematological problems including TTP. Thienopyridines are extensively and rapidly absorbed from the gut. CYP2C19 is a key enzyme involved in the metabolism of clopidogrel and many other drugs. Several studies have shown that CYP2C19activity is responsible for some of the variability in clopidogrel responsiveness amongst patients. Of interest is the finding that the prevalence of CYP2C19 defective genotypes varies with ethnic origin suggesting that differences in clinical efficacy of clopidogrel may in part be related to ethnic origin. |
| Prasugrel | Binds to the P2Y12 receptor preventing ADP-induced platelet aggregation. | The VerifyNow system is a whole blood method that has been evaluated as a rapid method for establishing the anti-platelet efficacy of Prasugrel therapy. | Prasugrel is a prodrug that is converted into the active metabolite by intestinal and hepatic Cytochrome P450 enzymes. Prasugrel has a greater effect that clopidogrel because it is metabolised more efficiently. |
| Dipyridamole | 1. Inhibits platelet phosphodiesterase which normally degrades cAMP. cAMP accumulates, intracellular Ca levels fall and this blocks the platelet response to ADP. 2. Thromboxane synthetase inhibitors therefore reduce the generation of Thromboxane A2 [TxA2] 3. Inhibits cellular update of adenosine into platelets and in addition it inhibits the breakdown of adenosine which leads to further accumulation of extracellular adenosine. |
Alpha T½ 40 minutes: Beta T½ 10 hours. | Dipyridamole is both a vasodilator through its effects on adenosine and an anti-platelet agent it is used primarily in combination with aspirin. |
| Abciximab | GpIIb/IIIa inhibitor. A chimeric human-murine antibody that target an epitope on the GpIIb/IIIa receptor close to a critical binding site for fibrinogen. | Abciximab has an Alpha T½ of 10 minutes and a Beta T½ 30 minutes although platelet aggregation may take 90-120 hours to return to normal even after the drug has been discontinued. The VerifyNow system is a whole blood method that has been evaluated as a rapid method for establishing the anti-platelet efficacy of GpIIb/IIIa antagonists. |
Thrombocytopaenia is a rare but serious side-effect of treatment with Abciximab and occurs in 0.3-0.6% of patients following their first exposure to Abciximab but in some patients up to 2.5%. The risk of thrombocytopaenia increases with subsequent exposures. Most patients who do develop the thrombocytopaenia associated with Abciximab do so shortly after administration [with a few hours of starting treatment] but a few do not do so until several days [5-8 days] after exposure. The mechanism is unclear but presumed to be antibody mediated and reflects the chimeric nature of the drug but also the target effect of Abciximab. |
| Tirofiban | GpIIb/IIIa inhibitor | T½ 2 hours The VerifyNow system is a whole blood method that has been evaluated as a rapid method for establishing the anti-platelet efficacy of GpIIb/IIIa antagonists |
Thrombocytopaenia has been reported in 0.5% of patients receiving oral Tirofiban. |
| Eptifibatide | GpIIb/IIIa inhibitor | T½ 2.5 hours |
|
| Ticagrelor | Binds [reversibly] to the ADP [P2Y12] receptor on platelets but is an active drug rather than a pro-drug and so does not require metabolic activation. |
T½ 6-13 hours | Administered orally and undergoes enzymatic degradation to generate active metabolite[s]. Ticagrelor achieves ~50-60% inhibition of ADP-induced platelet aggregation and appears more effective at achieving this than clopidogrel. |
| Cangrelor | Cangrelor is a direct competitive, reversible inhibitor of the ADP [P2Y12] receptor on platelets but is an active drug rather than a pro-drug and so does not require metabolic activation | T½ 9 minutes and so requires to be given by continuous iv infusion. Platelet function returns to normal within 60 minutes of discontinuation of the drug. | |
| Elinogrel | A reversible ADP receptor inhibitor. | ||
| Vorapaxar | Vorapaxar is a potent and specific competitive inhibitor of PAR-1. It has a T½ 126-269 hours and inhibitors TRAP-induced platelet aggregation for up to 4 weeks. The drug appears to dissociate slowly from its receptors and which may in part explain the long T½. | PAR-1 [Protease-Activated Receptor] belongs to a family of G-protein coupled receptors that are activated by proteolytic cleavage. Human platelets express both PAR-1 and PAR-4 receptors both of which can be activated by thrombin to induce platelet aggregation. Although activation of either receptor can induce platelet aggregation, the affinity for PAR-1 is 40X high than that of PAR-4 and therefore, the PAR-1 receptor is considered the major thrombin receptor on the surface of platelets. PAR-1 is also found on other cells e.g. endothelial cells - and activation of the PAR-1 receptor on these other cells may, in part account for some of the pro-inflammatory and proliferative effects of thrombin. |
|
| Atopaxar | Atopaxar is a potent and specific competitive inhibitor of PAR-1. It has a T½ 23 hours. | PAR-1 [Protease-Activated Receptor] belongs to a family of G-Protein coupled receptors that are activated by proteolytic cleavage. Human platelets express both PAR-1 and PAR-4 receptors both of which can be activated by thrombin to induce platelet aggregation. Although activation of either receptor can induce platelet aggregation, the affinity for PAR-1 is 40X high than that of PAR-4 and therefore, the PAR-1 receptor is considered the major thrombin receptor on the surface of platelets. PAR-1 is also found on other cells e.g. endothelial cells - and activation of the PAR-1 receptor on these other cells may, in part account for some of the pro-inflammatory and proliferative effects of thrombin. |
|
| Anti-Fibrinolytic Drugs | |||
| Tranexamic Acid | Tranexamic Acid [TA] is a synthetic lysine derivative that competitively inhibits the activation of plasminogen to plasmin by binding to and blocking the high affinity lysine binding sites on plasminogen. | Not necessary but fibrinolysis can be monitored with the TEG and ROTEM devices | TA binds to and blocks the high affinity lysine binding sites on plasminogen [and subsequently plasmin when plasminogen is activated] preventing the binding of plasmin and plasminogen to the fibrin clot or to fibrin monomers. Human plasminogen contains 4 to 5 lysine binding sites with low affinity for tranexamic acid and 1 with high affinity and it is the high affinity site of plasminogen that is involved in its binding to fibrin. Tranexamic acid is excreted in the urine and its use is contraindicated in patients with haematuria. TA has a of ~3.1 hrs. It is approximately 8-10X more active than EACA. |
| EACA | Similar to Tranexamic Acid | Not necessary but fibrinolysis can be monitored with the TEG and ROTEM devices | EACA has a similar mode of action to TA. |
| Vasopressin Analogues | |||
| DDAVP | DDAVP is a synthetic vasopressin analogue which, in a dose dependent manner increases FVIII and VWF release from endothelial cells. | 1. Monitoring of FVIII and VWF levels. 2. Fibrinolytic response if used in the DDAVP stimulation test. 3. Serum sodium and fluid intake. |
DDAVP is a synthetic vasopressin analogue which, in a dose dependent manner increases FVIII and VWF release from endothelial cells. DDAVP also causes the release of T-PA from endothelial cells and forms the basis for a test similar to the venous occlusion test, known as the DDAVP stimulation test. DDAVP is a potent anti-diuretic nonapeptide and fluid retention leading to hyponatraemia is a recognised side-effect. For these reasons, DDAVP should not be administered more than once every 24 hours. DDAVP also exhibits tachyphylaxis that is a reduction in the amount of FVIII and VWF that is released from endothelial cells with repeat dosing. |
| Direct Thrombin [IIa] Inhibitors | |||
| Hirudin | Hirudin is a naturally anticoagulant peptide that is derived from the saliva of the medicinal leech - Hirudo medicinalis. It is a direct thrombin inhibitor i.e. its anticoagulant activity is independent of Antithrombin | 1. APTT 2. Ecarin Clotting Time [ECT] 3. Thrombin Time 4. ELISA 5. Chromogenic Assays 6. The Hemoclot® Thrombin Inhibitor assay appears suitable for monitoring Direct Thrombin Inhibitors. |
Hirudin is a naturally anticoagulant peptide that is derived from the saliva of the medicinal leech - Hirudo medicinalis. Hirudin, in contrast to the Heparins, is a direct thrombin inhibitor and so its anticoagulant activity is independent of Antithrombin The therapeutic range for Hirudin is 0.5-2.5 µg/mL. Approximately 10% of patients who receive Hirudin develop anti-hirudin antibodies which prolongs its T½ and so increases its anticoagulant activity. |
| Lepirudin | Lepirudin is an almost identical recombinant form of Hirudin. | 1. APTT 2. Ecarin Clotting Time [ECT] 3. Thrombin Time 4. ELISA 5. Chromogenic Assays |
Lepirudin is an almost identical recombinant form of Hirudin. It has a T½ of ~1.3 hours and is given by iv infusion of subcutaneously. Lepirudin is used in the management of patients with HIT and in whom Heparins are contraindicated. In the EU, production of Lepirudin ceased on 1st April 2012. |
| Bivalirudin | Bivalirudin is a short synthetic peptide that is a potent, reversible, direct inhibitor of thrombin. | 1. APTT 2. Ecarin Clotting Time [ECT] 3. ACT 4. Chromogenic Assays |
Bivalirudin is a short [20 amino acid] synthetic peptide that is a potent, reversible, direct inhibitor of thrombin. Bivalirudin binds to both the catalytic site and the anion binding exosite of both circulating and clot-bound thrombin. This reaction is reversible as thrombin slowly cleaves the Bivalirudin. It has a T½ of ~25 minutes if renal function is normal. |
| Dabigatran etexilate [- see also expanded section below] |
Dabigatran is an oral, direct thrombin inhibitor with a high affinity for thrombin with reversible binding. | The Ecarin Clotting Time [ECT] directly assess the activity of thrombin in a plasma sample and displays a linear dose-response to therapeutic concentrations of Dabigatran. The APTT response curve flattens as the concentration of Dabigatran increases and is, therefore, relatively insensitive to the plasma concentrations of Dabigatran that are likely to be encountered in clinical practice. The PT is relatively insensitive to the plasma concentrations of Dabigatran. The Thrombin Time is very sensitive to the effects of Dabigatran and displays a linear dose-response curve over therapeutic levels but at high concentrations the actual clotting time may exceed the time that many instruments allow. The Hemoclot® Thrombin Inhibitor assay appears suitable for monitoring Direct Thrombin Inhibitors. |
Dabigatran is an oral, direct thrombin inhibitor with a high affinity for thrombin with reversible binding. Dabigatran etexilate is a prodrug that is converted into the active metabolite dabigatran with a low bioavailability. The intestinal absorption of Dabigatran etexilate is pH sensitive and its absorption is decreased in the presence of proton pump inhibitors [PPIs.] Dabigatran has a T½ of 12-17 hours with 80% of the drug excreted renally. Its metabolism is independent of cytochrome P450. |
| Argatroban | Argatroban is a direct thrombin inhibitor that is administered intravenously. | 1. APTT 2. The Hemoclot® Thrombin Inhibitor assay appears suitable for monitoring Direct Thrombin Inhibitors. |
Argatroban is a direct thrombin inhibitor that is administered intravenously. It is licensed for the treatment of patients with Heparin Induced Thrombocytopaenia [HIT]. Argatroban is metabolised in the liver and has a T½ of ~50 minutes. Argatroban is metabolised by the liver. The use of argatroban in patients who are also on on a VKA e.g. warfarin can lead to a falsely elevated PT and INR. In such cases performing a chromogenic FX assay can be useful in guiding warfarin dosage. |
| Defibrinating Agents | |||
| Ancrod | Ancrod is a defibrinating agent that acts by breaking down fibrinogen although the high levels of FDPs generated also act as an anticoagulant. Ancrod also appears to simulate fibrinolysis. Ancrod is rarely used today - originally it was used in cases of HIT or in patients with peripheral vascular disease where the use of Ancrod by reducing fibrinogen levels, decreased whole blood viscosity and potentially improved blood flow. |
Fibrinogen and Thrombin Time | Ancrod is a defibrinating agent isolated from the venom of the Malayan pit viper [Agkistrodon rhodostoma]. Ancrod has a T½ of 3-5 hours and the drug is cleared primarily through the kidneys. Ancrod cleaves fibrinogen so reducing fibrinogen levels leading to a reduction in plasma viscosity. |
| Factor Xa Inhibitors | |||
| Fondaparinux | See above under heparins | ||
| Idraparinux | See above under heparins | ||
| Rivaroxaban [- see also expanded section below] |
Rivaroxaban is an oral factor Xa inhibitor with competitive but reversible binding to Xa | Anti-FXa assay with Rivaroxaban calibrator. | Rivaroxaban is an oral factor Xa inhibitor with a competitive and reversible binding to Xa. There is some evidence that Rivaroxaban may bind not only to free FXa but also to Xa in the prothrombinase complex. The T½ of Rivaroxaban is 5-9 hrs. The drug is cleared both through the liver [2/3] and the kidneys [1/3] and so will accumulate in patients with liver and/or renal disease. |
| Apixaban | Apixaban is an oral factor Xa inhibitor with a competitive and reversible binding to Xa | Anti-FXa assay with Apixaban calibrator | Rivaroxaban is an oral factor Xa inhibitor with a competitive and reversible finding to Xa. The T½ of Apixaban is 9-14 hrs. As with Rivaroxaban, the drug is metabolised in the liver by the cytochrome P450 system and ~25% of the drug is cleared through the kidneys and the remainder by the liver. |
| Edoxaban | Oral direct FXa inhibitor | Anti-FXa assay with Edoxaban calibrator | Edoxaban is an oral factor Xa inhibitor with a T½ of 9-11 hrs. Edoxaban is eliminated both through the kidneys [35%] and liver [65%]. |
| Betrixaban | Oral direct FXa inhibitor | Anti-FXa assay with Betrixaban calibrator | An oral direct Xa inhibitor which is cleared almost exclusively by the liver. Betrixaban has a T½ of ~19 hours. A plasma-derived and recombinant FXa, modified to lack catalytic and membrane binding activities, has been developed and shown in laboratory studies to reverse the anticoagulant activities of these small molecule FXa inhibitors and also the LMWHs |
| Fibrinolytic Activators | |||
| Streptokinase | Streptokinase [SK] is a an enzyme produced Group C (beta) haemolytic streptococci. It binds to human plasminogen activating it to plasmin. This is independent of fibrin and therefore, SK leads to systemic hyperfibrinolysis. | Routine monitoring is not necessary but fibrinogen, the PT and APTT should be measured to assess the degree of fibrinolytic activity. | Intravenous infusion of Streptokinase is followed by increased fibrinolytic activity, which decreases plasma fibrinogen levels for 24 to 36 hours. The hyperfibrinolytic effect disappears within a few hours after discontinuation, but but the high levels of FDPs and low fibrinogen levels can lead to a prolonged thrombin time for up to 24 hours. The use of SK can sometimes lead to the development of anti-SK antibodies. |
| t-PA | T-PA is human recombinant protein that leads to fibrinolysis only at the site of vascular injury. It does not, therefore lead to systemic hyperfibrinolysis | Routine monitoring is not necessary | |
| Reteplase | Reteplase is a recombinant, non-glycosylated form of human t-PA that is modified to contain only 357 of the original 527 amino acids. These modifications prolong its T½ to 13-16 minutes | Routine monitoring is not necessary | |
| Tenecteplase | Tenecteplase is a recombinant form of human t-PA that is modified at three separate sites to prolong its T½. The T½ is 20-24 minutes | Routine monitoring is not necessary | |
| Urokinase | Urokinase is a thrombolytic agent obtained from human neonatal kidney cells grown in tissue culture. Urokinase is administered by intravenous infusion and is rapidly cleared by the liver with a T½ of 12.6 ± 6.2 minutes and a distribution volume of 11.5 L. Small fractions of the administered dose are excreted in bile and urine. |
Routine monitoring is not necessary | |
| APSAC | APSAC is an abbreviation for Acylated Plasminogen Streptokinase Complex. APSAC is a thrombolytic agent prepared from Streptokinase and human plasminogen, the active site of which is acylated to block activation by other plasma proteins, while retaining fibrin-binding capacity. | Routine monitoring is not necessary | |
| Staphylokinase | Recombinant Staphylokinase is a activator of fibrinolysis that is specific for fibrin. A derivative of Staphylokinase [THR-174] that is less immunogenic is in clinical studies. |
Routine monitoring is not necessary | |
{kind=link}