Mutational Analysis - A Brief Introduction
Introduction
Gene mutation analysis is a fundamental part of the investigation and management of an individual and their family with an inherited bleeding or thrombotic disorder. The focus of this section is to highlight some of the common mutations seen in individuals with inherited bleeding disorders or disorders associated with an increased risk of thrombosis. However the mutations, with some exceptions, are common to all genetic diseases.
The Clinical Molecular Genetics Society (CMGS) website provides a number of extremely useful guidelines including Best Practice Guidelines for molecular analysis in Haemophilia A, Haemophilia B, Von Willebrands Disease [VWD] together with practice guidelines for DNA sequence analysis and its interpretation.
The following is a brief list of the types of mutations that can occur in all genetic disorders:
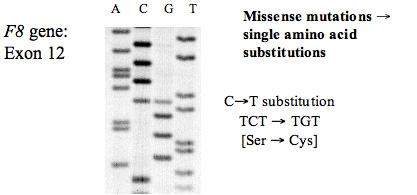
A. Missense Mutations: Missense mutations arise from single base pair [bp] substitutions that cause a change in the amino acid sequence. Some single bp mutations may not change the amino acid sequence This is because the amino acid code is degenerate – that is a single amino acid can be encoded by more than one codon.
In the example below the CG mutation [TCT→TGT] changes the Ser to a Cys and so alters the amino acid of the sequence. This is actually part of the gene for human Antithrombin SERPINC1]) and the gene for Antithrombin is located on the long arm of chromosome 1 [1q25.1]. As this is autosomally inherited, individuals can be heterozygous or homozygous for a mutation. In this case – the patient is heterozygous for the mutation and so shows at the arrow, two possible sequences: TCT or TGT.
This is an autoradiograph of an 35S sequencing gel. An electropherogram would show two peaks at the arrow representing the two possible sequences.
B. Nonsense Mutations: Nonsense mutations arise from single bp substitutions and which cause the creation of a premature stop codon. In the example below the C→A [TGC→TGA] mutation changes the Cys to a Stop codon.
Normal sequence
Ser
5’- ATG CTG TGCMutant sequence
Stop
5’- ATG CTG TGA
Some out-of-frame deletions or insertions may give rise to the creation of a premature stop codon downstream of the insertion/deletion.
C. Splice Mutations: Specific nucleotide sequences at the splice sites guide the correct splicing of DNA. If a mutation alters one of these signals, then part of or the whole intron may not be removed and remains as part of the final RNA molecule. The translation of this sequence alters the sequence of the protein product.
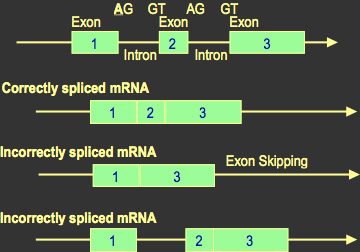
Some frameshift mutations can be partially corrected due to transcriptional stuttering and/or ribosomal slippage and this leads to the production of some normal protein.
D. Frameshift mutations: In a frameshift mutation, one or more bases are inserted or deleted. Because the DNA coding sequence in is based upon a series of triplet sequences or codons – deletions or insertions can be ‘in-frame’ or ‘out-of-frame’. An in-frame deletion must involve at least 3 DNA bases (it may be more and is usually multiple of 3) removes an entire codon and so may lead to the deletion of an amino acid from a protein e.g.
a. In-frame deletion
Wild-type sequence [part of the human Antithrombin gene]
Glu Leu Ser Lys Ala Asn
5’- GAA CTG TCC AAG GCC AAT
Mutant Sequence: in-frame 3bp [CTG] deletion
Glu Ser Lys Ala Asn
5’- GAA TCC AAG GCC AAT
Deletion of the CTG codon removes the leucine amino acid but does not change the reading frame and the other amino acids remain unchanged. However, whilst the reading frame is maintained, in-frame deletions give risk to a protein that lacks one or more amino acids. This frequently disturbs the tertiary structure of the protein and as a result it either fails to be secreted to if it does, it is rapidly removed from the circulation.
Of course a 3bp deletion could remove part of one codon and part of the next and give rise to an out-of frame deletion. Similarly a 3bp insertion could occur between the nucleotides of a codon leading to an out-of frame insertion.
b. Out-of-frame deletion
Mutant Sequence: in-frame 2bp out-of-frame deletion
In contrast an out-of-frame deletion changes the reading frame and gives rise to an amino acid sequence which following the deletion is completely different from the native sequence.
Wild type sequence
Glu Leu Ser Lys Ala Asn
5’- GAA CTG TCC AAG GCC AAT
In this sequence if we delete the CT in the CTG that encodes Leucine then this changes the reading frame and the corresponding amino amino acid sequence
Glu Val Gln Gly Gln ...
5’- GAA GTC CAA GGC CAA T..
Similar changes to the reading frame are seen with in-frame insertions (multiples of 3bp) or out-of frame insertions (more or less than 3bp but never multiples of 3bp).
E. Major Gene Deletions/Insertions: Major gene deletions or insertions result in the loss of one or more genes depending upon the size of the event see: http://en.wikipedia.org/wiki/File:Types-of-mutation.png – for a diagrammatic illustration of this.
Haemophilia B Leyden
Haemophilia B Leyden is a bleeding disorder characterized by an altered developmental expression of blood coagulation factor IX. Factor IX levels are low at birth but reach normal adult values at puberty. This form of Haemophilia B has been found to be associated with a variety of single point mutations in the F9 promoter region (-40 to -9) that contains overlapping binding sites for hepatocyte nuclear factor 4 (HNF-4) and androgen receptor.
Mutations Leading to FIX Deficiency During Anticoagulant Therapy
Bleeding complications in patients receiving oral anticoagulants are common but an unusual selective decrease in factor IX activity (1–3% of normal) has been observed in some patients receiving Warfarin. Subsequent molecular analyses has identified two different missense mutations within the F9 gene at alanine –10 [Ala to Val and Ala to Thr.]
Alanine –10 lies within the pro-peptide of the factor IX and at a position that is essential for binding of a vitamin K-dependent carboxylase that modifies glutamic acid residues in the GLA domain. Individuals carrying the F9 Ala –10 mutation have a decreased affinity for the carboxylase enzyme and in the presence of Warfarin and other vitamin K dependent anticoagulants show a significant fall in factor IX activity compared to the other vitamin K dependent clotting factors.
Intron 1 and Intron 22 Inversions within the F8 Gene
In severe haemophilia A, the Intron 22 inversion is the most prevalent mutation, accounting for about 40–50% of all mutations. The inversion results from a recombination between a 9.5-kb region in Intron 22 (int22h1) and either of two extragenic, distal homologues [located about 300 kb distal to the F8 gene at the telomeric end of the X chromosome.], int22h2 and int22h3; int22h2 and int22h3 are more than 99% identical to one another. Recombination produces an inversion because the extragenic homologues are in the opposite orientation relative to int22h1.
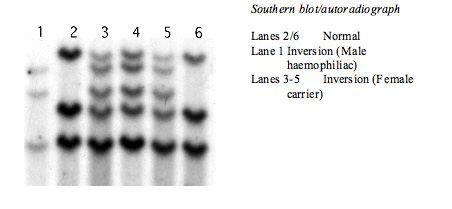
A second inversion within Intron 1 of the F8 gene exists although is significantly less common than the intron 22 inversion, with a prevalence of around 4.8% in the white population. This genomic rearrangement is the result of homologous recombination between two nearly identical 1041-base pair sequences, int1h-1 and int1h-2, in opposite orientations, positioned within Intron 1 of the F8 gene, resulting in the production of two chimeric mRNAs.
Whilst historically the Intron 22 inversion was detected by Southern Blotting - it is now screened for by PCR or NexGen sequencing.